多变量分析:肿瘤分类的关键
Josef Büulles, Stephen David, Stephanie Bourin
GE Healthcare, Discovery Systems, Amersham Biosciences AB, Uppsala, Sweden
|
简介
双向电泳使得在一块胶内观察上千个蛋白点以及它们的修饰成为可能。Ettan™2D DIGE 系统的引入大大消除了经典双向电泳中较高的系统偏差。尽管降低了系统误差,固有的生物个体差异也会掩盖诱导产生的生物学变异。因此,就需要更有效的数据分析方法去发现这些标记蛋白质。专为结合Ettan™ 2D DIGE应用开发的扩展数据分析(extended data analysis,EDA)模块,使得该方法的全部优势得以发挥。该模块在单变量分析的基础上加入了多变量分析,单变量分析功能现在已经存在于DeCyder™2D 分析软件中。
EDA将有助于发现表达模式,寻找表达谱类似的蛋白质以及鉴别的方法(发现可用于诊断和/或预后的疾病标记物)。最后,EDA可以将未知样本按已知的类别进行归类(例如肿瘤),这对于个性化的医疗具有更深层次的意义。
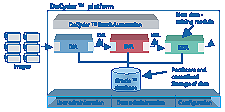 |
|
图1:DeCyder™2D 平台的分析流程 |
材料与方法
本文中使用的是由Peter James教授(University of Lund, Sweden)提供的人卵巢癌研究的DeCyder™ BVA 工作区。所有的病理活检材料按病理学分级分为下面的三类:正常,良性和恶性。使用了18个患者的标本(3例正常,4例良性,11例恶性)来完成后续的分析。所有的患者标本都分析了两次,使用正反标记和样品随机化的方法。
通过对10个未知的患者样本的分类来验证分析结果与病理学分类。
一些蛋白没有出现在所有的胶上,原因是由于样本质量不好或匹配误差。而几乎不需要花费时间来更正这些问题,证明了强大的统计学工具能够检测疾病标记物或是对未知的样本进行分类。接下来的分析中,我们选择了在所有的点图上出现率高于80%的蛋白点。通过计算1-wayANOVA的多重比较测试以及选择p值<0.001,我们将初始的3042个蛋白点降到171个。判别分析明确显示,剩下的171个蛋白点使我们可以区分已知的样本。
结果
为了初步了解该软件对于样本分类的有效程度,进行了PCA分析。下图(图2)中显示出对于不同类的样本进行了清晰的分类。
假定那些显示统计学最显著的表达差异的蛋白点可以被用来最好的区分不同的类别,选择那些p值<1E-11(图3)的蛋白点用来做聚类分析。结果表明这些蛋白点得不到预定的结果。而用整个171个蛋白的组群能够得到最佳的分类。
|
|
我们选择Pearson法测量距离来进行分级聚类,类似于连锁的方法。所有的恶性样本被聚类成簇而且和所有其它的样本完全分离开。一半良性样本和正常样本距离很近,而另一半则分开,但是仍然是所有的良性和正常的样本构成了各自的共同组群。
 |
|
图2:PCA图显示对于三种已知分类样本的聚类结果 |


|
|
图3:P<1E-11的蛋白的标准化丰度对数值.其中的四种蛋白可以区分良性/正常和恶性,介是公有一种可以区分良性和正常. |
分类:
选择“Regularized Discriminant Analysis”进行分类研究,已知的样本被用到自学运算法则中的培训部分。这些分类程序的目的是确认已知样本是否可以被正确分类以及未知样本是否能被分入已知的类别中。
结果表明所有已知的样本都被正确的分类,10个未知样本中的8个被正确的分到已知的类别,其中两个未知样本分类结果与前期的病理学分类不符合。一个恶性的被分类为良性,一个良性的被分类为恶性。
上述不符合的数据正在研究中。
结论
●
多变量统计分析对于双向电泳产生的蛋白表
达数据的分析是一个有效的手段
● EDA使得非专业的统计人员可以快速和有效
进行综合的多变量分析
● 对于双向电泳数据应用综合多变量统计分析
可以增强该技术在诊断上的能力
● EDA使得可以从双向电泳得到的数据中鉴定
出疾病标记物从而发展外在的诊断检测方法
|