
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
可逆单晶光化学与自旋态切换:七氰基钼酸盐的高温光磁开关新机制
【字体: 大 中 小 】 时间:2025年09月30日 来源:Nature Communications 15.7
编辑推荐:
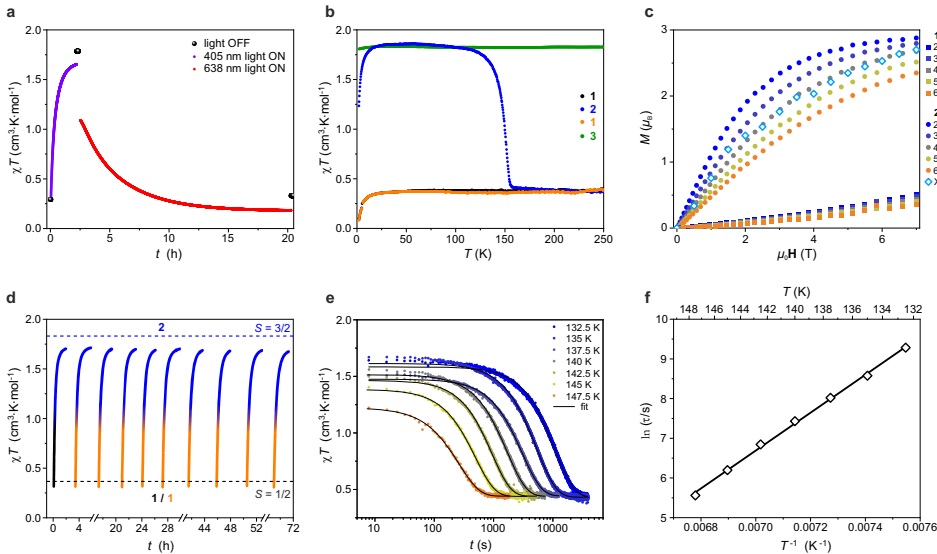
本研究针对固态光化学调控难题,报道了K4[MoIII(CN)7]・2H2O晶体中通过可见光触发配位键可逆断裂/重组,实现七配位低自旋(S=1/2)与六配位高自旋(S=3/2)态的可逆转换。该转化具有100%效率、波长选择性(405nm解离/638nm结合)和150K的创纪录高温稳定性,为开发室温光控磁性材料提供了新范式。

 生物通微信公众号
生物通微信公众号
知名企业招聘

