
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基于甘草酸靶向流感病毒血凝素的新型广谱抑制剂发现及其心脏安全性优化研究
【字体: 大 中 小 】 时间:2025年08月27日 来源:Virology Journal 3.8
编辑推荐:
本研究针对流感病毒A(IAV)广泛耐药性问题,通过结构-活性关系分析发现抗病毒药物Arbidol虽能靶向保守的HA融合肽区,但其hERG钾通道抑制特性(IC50=0.3030μM)存在心律失常风险。团队筛选50种天然产物,发现甘草酸(Glycyrrhetinic acid)与HA结合自由能(-5.26 kcal/mol)优于Arbidol,且柔性分子结构规避了hERG抑制,为开发安全广谱抗IAV药物提供新策略。
每年流感季来临,H1N1和H3N2等流感病毒A(IAV)亚型总能引发全球流行。尽管疫苗覆盖率逐年提升,2023-2024季保护率仅44%,而神经氨酸酶抑制剂(NA)的耐药性突变更让临床治疗雪上加霜。传统药物如奥司他韦(Oseltamivir)靶向易变异的NA蛋白,但病毒通过抗原漂移(antigenic shift)轻松逃逸。这迫使科学家将目光转向更保守的靶点――血凝素(HA)的茎部区(stem region),这里藏着病毒入侵细胞的"万能钥匙":融合肽(FP)。
正是在此背景下,Arbidol(Umifenovir)进入了研究者视野。这种含吲哚结构的广谱抗病毒药能嵌入HA的疏水腔,阻断酸性环境下的构象重排,从而抑制病毒膜融合。但令人担忧的是,其分子中的叔胺和芳香环恰是心脏hERG钾通道抑制剂的典型特征,可能引发QT间期延长甚至猝死。如何保留Arbidol的广谱抗病毒特性,又规避心脏毒性?北京工业大学生物工程学院Ran Yu团队在《Virology Journal》发表的研究给出了创新解决方案。
关键技术方法
研究采用多学科交叉策略:1) 基于HA(PDB ID:5THF)和hERG(PDB ID:5VA1)晶体结构的分子对接筛选50种天然产物;2) 自动膜片钳系统(QPatch 48X)定量检测化合物对hERG尾电流的抑制率;3) 结合自由能计算评估HA结合稳定性;4) 以阳性对照药Cisapride验证实验体系可靠性。
研究结果
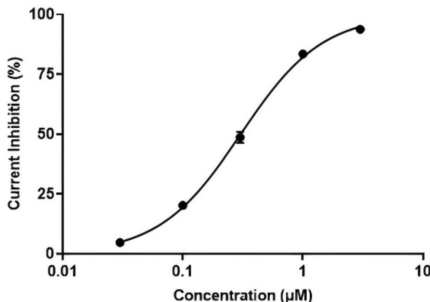
hERG抑制性验证
自动膜片钳显示Arbidol对hERG的IC50仅0.3030μM,3μM时抑制率达93.79%。分子对接揭示其通过π-π堆积与TYR-497/ALA-478结合,叔胺基与HIS-492形成氢键,完美契合hERG抑制剂特征。相比之下,奥司他韦和瑞德西韦(Remdesivir)因缺乏刚性共轭结构而无此毒性。
HA抑制剂筛选
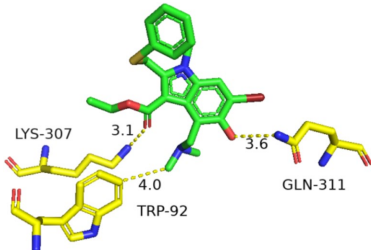
从《伤寒论》衍生的天然产物库中,甘草酸与HA结合能(-5.26 kcal/mol)优于Arbidol(-5.02 kcal/mol),通过三个氢键锚定LYS-307/310等关键残基。其五环三萜结构因柔性优势,既维持HA结合力,又避免与hERG疏水腔嵌合。伪麻黄碱虽结合位点相似,但苯环结构仍存hERG风险。
结论与意义
该研究首次系统评估Arbidol的hERG毒性机制,并基于结构优化策略发现甘草酸这一安全替代物。其意义在于:1) 确立HA茎部区为广谱抗病毒设计的黄金靶点;2) 提出"柔性分子规避hERG"的新设计原则;3) 为传统中药活性成分改造提供范例。未来研究需在动物模型验证甘草酸的体内抗病毒效力,但其已展现出超越现有药物的临床转化潜力。
生物通微信公众号
知名企业招聘

