
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基因组空间的几何构建:基于自然向量的全生物宇宙模型与凸包原理验证
【字体: 大 中 小 】 时间:2025年08月05日 来源:The Innovation 33.2
编辑推荐:
本研究通过整合来自细菌、古菌、真菌等七大类生物的可信参考序列,构建了"全生物宇宙"模型,利用自然向量方法验证了多层级凸包分离原理。研究发现凸包分离源于生物学特性而非高维空间数学特性,并开发了适用于分类的最优度量标准,为基因组空间几何结构提供了理论支撑,对生物序列分类和未知序列预测具有重要价值。
在探索生命奥秘的征程中,如何从数学角度解析基因组间的几何关系一直是生物信息学的核心挑战。传统序列比对方法虽然精确但计算复杂,而现有无比对方法又难以满足基因组空间构建的数学严谨性要求。尤其当面对海量生物数据时,亟需建立统一的几何框架来揭示不同生物类群间的本质差异。
清华大学数学科学系的研究团队在《The Innovation》发表突破性研究,通过整合NCBI数据库中7大类30,121条可靠序列,首次构建了完整的"全生物宇宙"模型。该研究采用自然向量方法(将DNA序列转化为k-mer统计矩向量),在68维空间中验证了贯穿生物分类层级的凸包分离现象:不仅细菌、古菌等七大"生物星系"的凸包互不相交,其内部各科级分类单元也呈现同样规律。更关键的是,研究证实这种分离源于生物学特性而非高维数学特性――通过标签随机化实验获得97-100%的显著比。基于此,团队开发了整合k-mer信息的自然度量标准,在七大生物类群中实现82.2-92.7%的分类准确率,并成功应用于硅藻类争议物种的系统发育分析。
关键技术包括:1)从NCBI获取七大类生物基因组数据并严格过滤;2)构建1-mer和k-mer自然向量空间;3)采用优化算法验证凸包分离性;4)设计7种加权策略的L1/L2自然度量;5)结合t-SNE降维可视化。
主要结果:
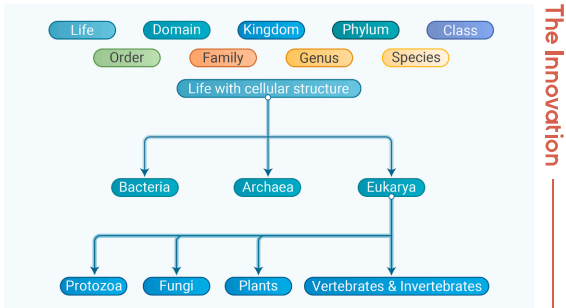
全生物宇宙概览
建立包含细菌、古菌等七大类生物的层级化向量空间,类比宇宙-星系-星簇结构。
凸包原理验证
在68维1-mer空间中,所有生物星系及其内部科级单元(古菌为门级)的凸包均严格分离,所需维度远低于理论预期。
自然度量构建
最优度量因生物类群而异,如细菌采用9-mer L1距离(90.3%准确率),而原生生物适用Σ(1/k1.5)dk1组合(92.7%准确率)。
系统发育应用
基于线粒体序列的自然度量分析,支持骨骼藻科(Skeletonemataceae)与硅藻科(Bacillariaceae)应同属硅藻纲,为分类争议提供分子证据。
体系稳定性验证
新增170条植物染色体数据后,凸包分离维度与分类精度保持稳定(82.4% vs 82.2%)。
这项研究首次在全生物尺度验证了凸包原理的普适性,其创新性体现在:1)建立首个整合所有生物类群的几何框架;2)揭示凸包分离的生物学驱动本质;3)开发可适应不同类群特性的动态度量标准。该成果不仅为DARPA提出的"基因组空间几何"难题提供解决方案,更启示可通过逆向工程在凸包内预测未知序列,为合成生物学和物种发现开辟新途径。正如研究者所述:"全生物宇宙中,七大星系与无数星簇的分离模式,恰似被生命密码扭曲的时空度量。"
生物通微信公众号
知名企业招聘

