
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:肌强直性营养不良的分子遗传学及治疗方法的演变
【字体: 大 中 小 】 时间:2025年07月04日 来源:Journal of Human Genetics 2.6
编辑推荐:
这篇综述系统阐述了肌强直性营养不良(DM1/DM2)的分子机制与治疗进展,重点解析了非编码区CTG/CCTG重复扩增导致的RNA毒性(如核RNA灶形成、MBNL蛋白 sequestration)及其引发的剪接失调(如CLCN1、SCN5A等基因异常)。文章详述了靶向RNA的核酸药物(如ASOs、RNAi)和小分子(如GSK3β抑制剂 Tideglusib)的临床转化潜力,并探讨了CRISPR和R-loop调控等新兴策略。多组学证据揭示了DM作为多系统疾病的复杂性,涉及衰老(p16/p53通路)、代谢(AMPK)和神经退行性(RAN翻译)等交叉机制。
肌强直性营养不良(DM)作为最常见的成人型肌营养不良症,以骨骼肌症状(肌强直、进行性肌萎缩)和多系统受累为特征。DM1由DMPK基因3'非翻译区CTG重复扩增(>50次)引发,而DM2源于CNBP基因内含子1的CCTG扩增(>75次)。新生儿筛查显示DM1实际发病率可能高达1/1786,远超既往认知。临床分型包括先天型、青少年型、成人型和晚发型,典型表现为"斧头脸"面容、远端肌无力及心脏传导异常。多系统症状涵盖认知障碍(前额叶功能受损)、胰岛素抵抗、白内障等,其严重程度常超越肌肉症状本身影响患者生存质量。
DM1的CTG重复呈现显著的体细胞不稳定性:白细胞中重复数约数百次,而肌肉/脑组织可达数千次。这种动态突变源于DNA滑移链结构形成和错配修复异常,其中转录介导的R-loop结构(CUG RNA与CAG DNA链杂交体)是关键驱动因素。值得注意的是,约3-11%患者携带CCG/CTC等重复中断序列,这类变异与较轻表型相关。先天性DM1中DMPK侧翼区CpG高甲基化与疾病严重度正相关,提示表观遗传调控的重要性。母系遗传时CTG重复呈现显著"遗传早现"现象,而父系传递更易发生重复收缩。
致病核心在于突变RNA的毒性功能获得:
RNA灶与剪接失调:含CUG/CCUG重复的异常RNA在核内形成稳定发夹结构,捕获肌肉盲样蛋白(MBNL1/2),同时激活CUGBP Elav样因子1(CELF1)的磷酸化稳定。这种MBNL功能缺失与CELF1过表达导致400余个基因剪接异常,例如:
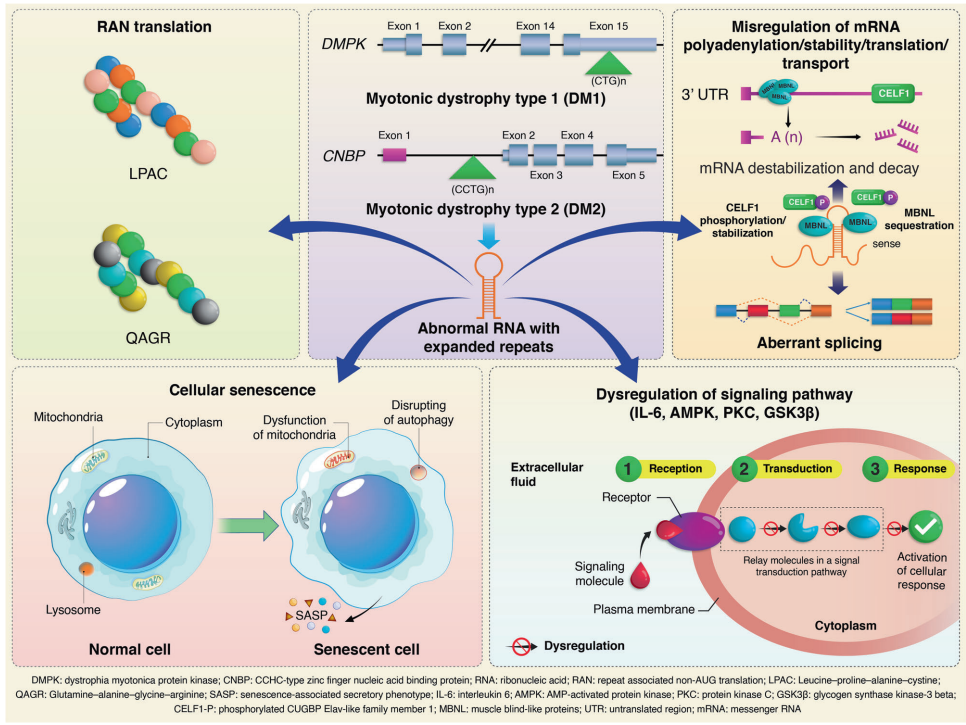
非经典翻译与信号通路紊乱:
加速衰老假说:DM1细胞呈现p21/p16高表达、线粒体功能障碍等衰老标志。动物模型证实senolytics(衰老细胞清除剂)可缓解表型,支持DM作为早衰综合征的新认知。
临床策略聚焦于消除毒性RNA或阻断其下游效应:
核酸药物:
小分子药物:
基因编辑:
DM的病理机制阐释为RNA介导疾病的范式研究提供了经典模型。尽管核酸药物在组织递送效率上仍存挑战,多靶点联合干预(如ASO+senolytics)可能成为未来方向。随着对体细胞不稳定性机制的深入,直接调控重复扩增的"治本"策略或将开启DM治疗新时代。
生物通微信公众号
知名企业招聘

