
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
西伯利亚芫菁(Mylabris sibirica)染色体水平基因组组装:为害虫防控与药用开发提供关键遗传资源
【字体: 大 中 小 】 时间:2025年02月16日 来源:Scientific Data 5.8
编辑推荐:
本研究针对危害油菜生产的西伯利亚芫菁(Mylabris sibirica)缺乏基因组资源的现状,通过PacBio、Illumina和Hi-C技术构建了首个染色体水平基因组(138.45 Mb,N50=13.84 Mb),注释到11,687个蛋白编码基因和35.46%重复序列。该成果为解析其发育调控、斑蝥素(Cantharidin)合成机制及RNAi靶标筛选奠定基础,兼具农业害虫防控与抗癌药物开发双重价值。
在中国青海省海东市的油菜田中,一种名为西伯利亚芫菁(Mylabris sibirica)的小甲虫正悄然成为农业生产的隐形杀手。这种隶属于芫菁科(Meloidae)的超变态昆虫,其成虫阶段专门取食油菜(Brassica napus)花朵,每年造成重大经济损失。更令人惊奇的是,这种害虫体内合成的斑蝥素(Cantharidin)已被证实对肝癌、胃癌等多种恶性肿瘤具有显著疗效,但其分子机制始终笼罩在迷雾中。由于缺乏高质量的基因组资源,科学家们既难以开发针对性的害虫防控策略,也无法深入挖掘其药用价值,形成了农业与医学研究的双重瓶颈。
中国农业科学院油料作物研究所农业农村部农业转基因生物溯源重点实验室的研究团队决心破解这一难题。他们采用多组学技术首次构建了西伯利亚芫菁染色体水平基因组,相关成果发表于《Scientific Data》。这项研究不仅填补了芫菁科昆虫基因组空白,更搭建起连接农业害虫防控与抗癌药物开发的分子桥梁。
研究团队运用PacBio HiFi长读长测序(112X)、Illumina短读长(201X)和Hi-C染色体构象捕获(151X)技术,结合RNA-seq数据,通过Hifiasm和3D-DNA等算法完成基因组组装。样本采自青海油菜田的雄性成虫,经肠道剔除后用于DNA/RNA提取。通过k-mer分析预估基因组大小为180.22 Mb,最终获得138.45 Mb的高质量组装,其中99.85%序列锚定到10条假染色体上,包含11,687个预测基因。
西伯利亚芫菁作为重要的油菜害虫和药用资源昆虫,其基因组资源长期匮乏。本研究成功构建首个染色体水平参考基因组,BUSCO评估显示100%完整性(1,358个单拷贝基因),重复序列占比35.46%,为后续功能研究提供基础。
研究采用多平台测序策略,通过Hi-C技术将22个contig锚定到10条染色体,X染色体通过测序深度差异(18.48X)被成功鉴定。Merqury评估显示组装质量值(QV)达59.336,Illumina数据比对率95.19%,验证了组装的准确性。
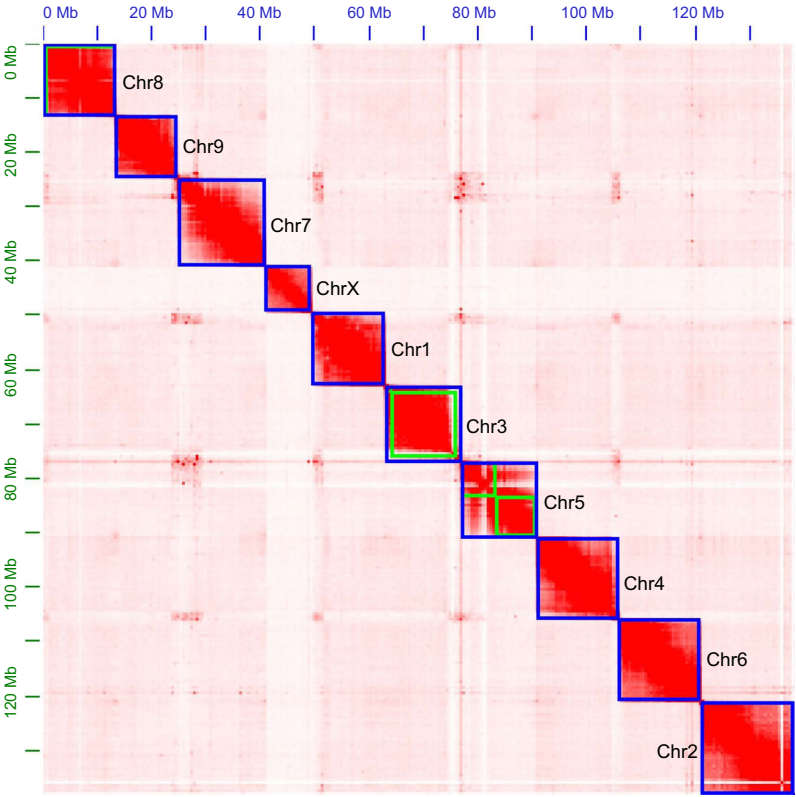
基因组注释揭示转座元件占35.46%,其中DNA转座子(4.21%)、LINEs(1.07%)和LTRs(1.60%)为主要类型。circos图显示基因密度与重复元件分布特征,X染色体(8.47 Mb)显著小于常染色体(11.35-16.80 Mb)。
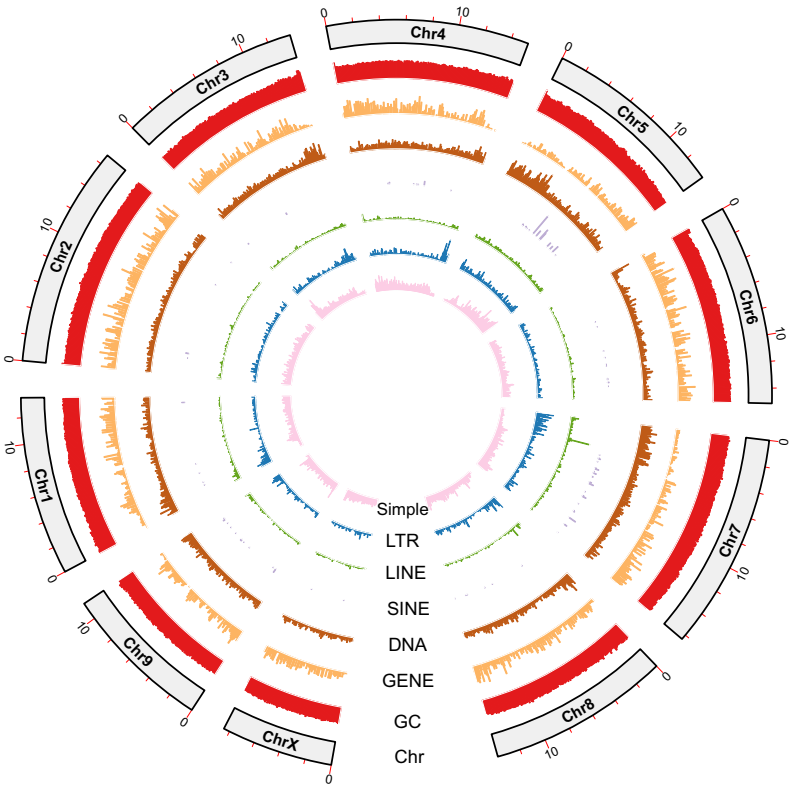
预测基因平均含5.9个外显子,平均长度5,959.6 bp。通过InterProScan和eggNOG注释到9,253个GO术语、4,403条KEGG通路,其中与斑蝥素合成相关的细胞色素P450和倍半萜合成酶基因家族值得关注。
这项研究创建的基因组资源具有多重意义:在农业领域,为开发基于RNAi的靶向防控技术提供分子靶标;在医药领域,为解析斑蝥素生物合成通路创造条件;在进化生物学方面,其Xyp性别决定系统为研究鞘翅目性染色体演化提供新模型。研究者特别指出,该基因组将加速"以虫治虫"策略开发――通过调控斑蝥素合成基因既可增强其作为天敌引诱剂的效能,又能降低其对作物的直接危害,实现农业害虫管理的生态平衡。
生物通微信公众号
知名企业招聘

