内质网蛋白CLCC1介导疱疹病毒与宿主核膜融合的新机制及其在核孔复合体形成中的作用
《Nature Communications》:ER protein CLCC1 promotes nuclear envelope fusion in herpesviral and host processes
【字体:
大
中
小
】
时间:2025年11月22日
来源:Nature Communications 15.7
编辑推荐:
本文推荐一项关于疱疹病毒核逃逸机制的重要研究。为解决疱疹病毒如何突破核膜这一关键问题,研究人员通过全基因组CRISPR筛选发现内质网蛋白CLCC1是促进核膜融合阶段的关键宿主因子。研究证实CLCC1缺失会导致核周包膜病毒颗粒(PEVs)积累,并发现疱疹病毒通过水平基因转移获得CLCC1同源基因。该研究不仅揭示了病毒利用宿主机制完成核逃逸的新途径,还发现CLCC1在核孔复合体形成等细胞过程中的重要作用,为开发广谱抗疱疹病毒疗法提供了新靶点。
在病毒与宿主持续博弈的漫长进化史上,疱疹病毒目(Herpesvirales)成员发展出了一套独特的核逃逸策略。这些病毒在细胞核内完成基因组复制和衣壳组装后,面临着一个关键挑战:如何将体积庞大的衣壳(直径约125纳米)穿过核膜屏障运送到细胞质中继续成熟。与传统病毒通过核孔复合体(NPC)的核转运方式不同,疱疹病毒采用了一种非常规的核逃逸途径:首先在核内膜(INM)出芽形成核周包膜病毒颗粒(PEVs),然后这些临时包膜与外核膜(ONM)融合,将无包膜衣壳释放到细胞质中。
虽然病毒编码的核外膜复合体(NEC)介导的出芽阶段已被广泛研究,但融合阶段的分子机制数十年来一直是个谜团。研究人员曾推测病毒包膜糖蛋白gB和gH可能参与此过程,然而基因敲除实验显示这些蛋白的缺失仅引起轻微表型,提示可能存在宿主因子参与。与此同时,细胞自身也存在类似的核膜融合过程,如核孔复合体插入、核膜出芽(NEB)用于输出大分子mRNA/蛋白复合物或错误折叠蛋白,但这些过程的融合蛋白尚未确定。
为破解这一谜题,研究团队开发了一种基于流式细胞术的定量检测方法,用于测量HSV-1的核逃逸效率。通过将表达Cas9的HeLa细胞与全基因组CRISPR sgRNA文库(Gattinara库,约40,000个sgRNA)结合,他们进行了大规模筛选,意外发现内质网蛋白CLCC1是核逃逸的最主要正调控因子。
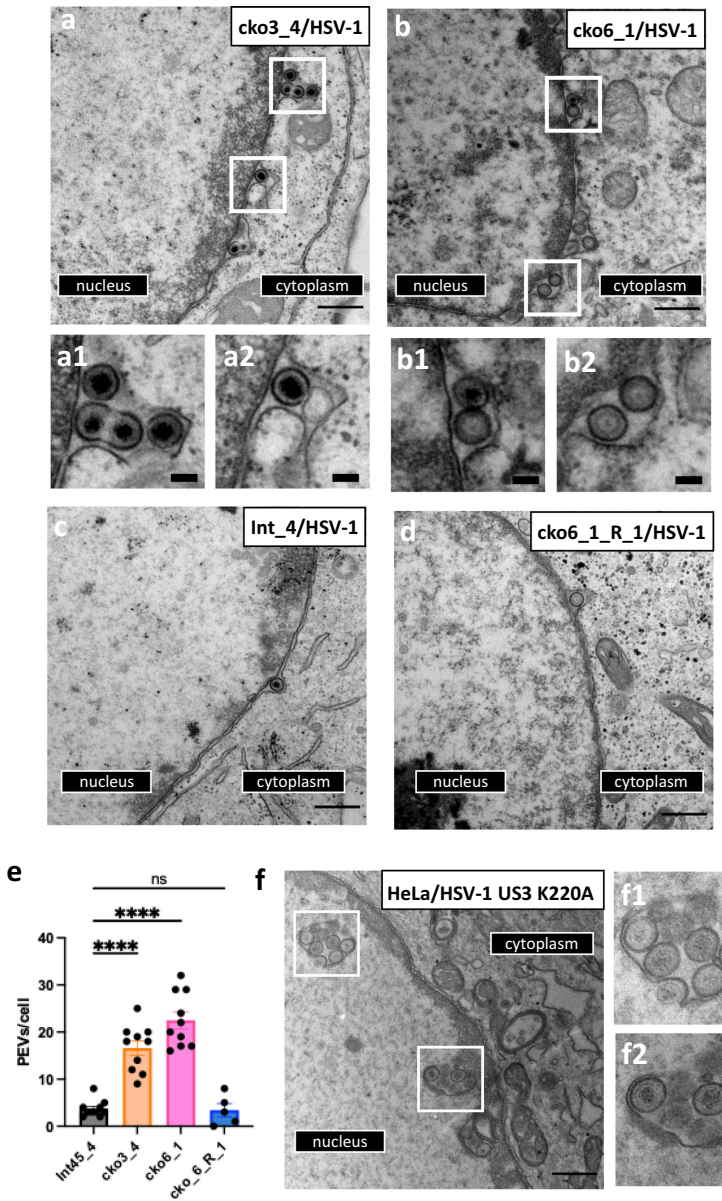
验证实验表明,在CLCC1敲除(KO)细胞系中,HSV-1核逃逸效率显著降低,透射电子显微镜(TEM)观察发现PEVs在核周间隙(PNS)大量积累,表明融合阶段受阻。更重要的是,这种现象不仅存在于HSV-1感染,在HSV-2和伪狂犬病毒(PRV)中同样出现,说明CLCC1在α疱疹病毒亚科中具有保守功能。
0.05 (p values from left to right 0.1653 and >0.9999). Significance was calculated using one-way ANOVA, with multiple comparisons, n=5 for Int_4 and cko6_1_R_1, n=4 for cko3_4 and cko6_1. Source data for graphs are provided in Source Data 9.'>
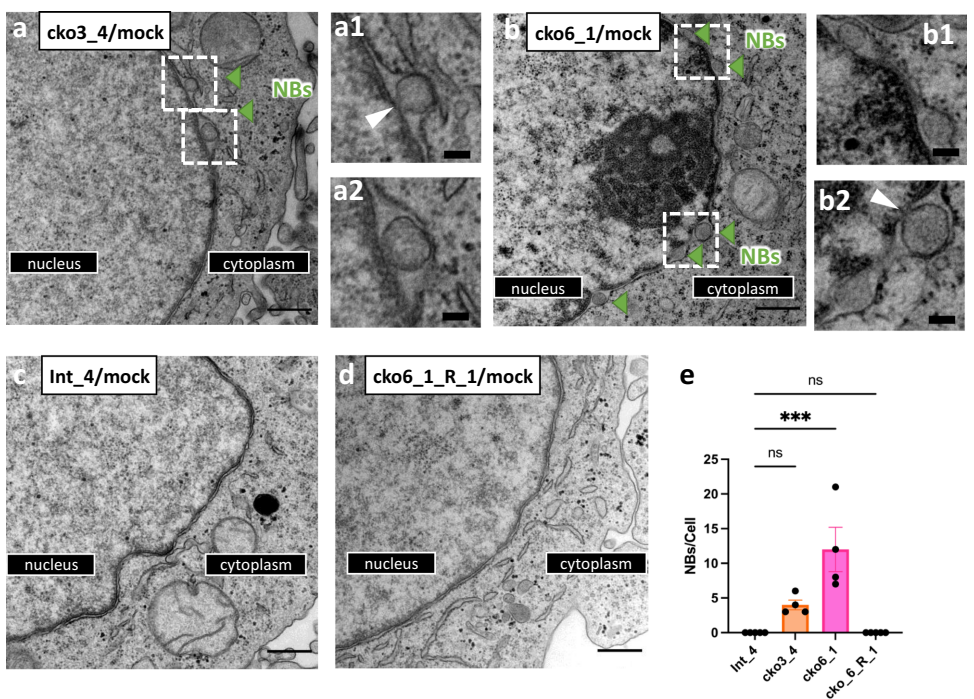
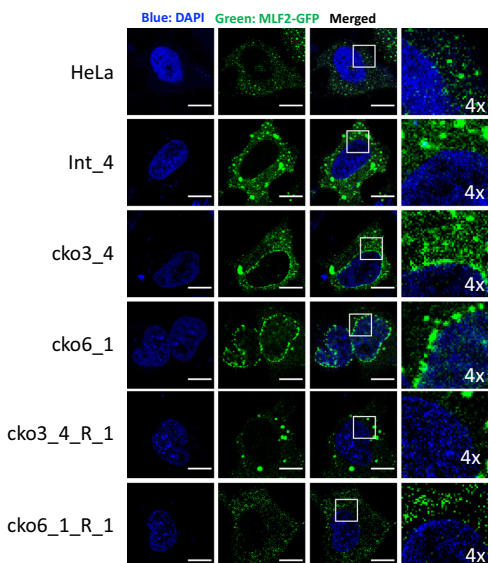
尤为引人注目的是,在未感染的CLCC1-KO细胞中,研究人员观察到了核膜出泡(NBs)现象,这与之前报道的Torsin ATP酶缺失表型相似,表明CLCC1在正常细胞核膜形态发生中同样发挥重要作用。髓系白血病因子2(MLF2)定位实验进一步证实,CLCC1缺失导致MLF2在核膜上形成点状聚集,提示核孔复合体插入或核输出存在缺陷。
系统进化分析揭示了另一个惊人发现:感染软体动物的Malacoherpesviridae和感染鱼类的Alloherpesviridae基因组中编码有CLCC1同源基因(vCLCC1),这些基因通过水平基因转移(HGT)从宿主基因组中获得,且独立发生了三次。病毒CLCC1同源蛋白与细胞CLCC1(cCLCC1)在核心区域高度保守,特别是形成二硫键的两个半胱氨酸(C254和C279)和多个关键氨基酸残基。
突变拯救实验显示,与氯离子通道活性相关的突变(S263R和W267R)仍能部分或完全拯救核逃逸缺陷,而影响PIP2(磷脂酰肌醇4,5-二磷酸)互作的K298E突变则完全丧失拯救能力。特别值得注意的是,高度保守的D277R突变不仅无法拯救CLCC1-KO细胞的核逃逸缺陷,还在表达内源CLCC1的细胞中表现出显性负效应,提示该残基可能位于CLCC1寡聚化界面。
AlphaFold结构预测表明,CLCC1可形成六聚体、十聚体甚至十六聚体,其中D277被预测参与形成分子间盐桥。结构模型显示CLCC1具有三个跨膜螺旋(TM1-3)、一个ER腔侧的拳状结构域(FD)和一个长的两性螺旋(AH)。特别值得注意的是,FDh2(拳状结构域的"指节"部分)富含芳香族氨基酸(W260、W265、F268和W272),类似于病毒融合蛋白的融合肽,且其构象在寡聚化时发生改变,提示可能直接参与膜融合过程。
本研究运用了全基因组CRISPR-Cas9筛选技术鉴定HSV-1核逃逸关键宿主因子;建立了流式细胞术核逃逸定量分析方法;通过透射电子显微镜(TEM)观察核膜超微结构变化;采用系统进化和基因组学分析病毒CLCC1同源基因的起源;利用AlphaFold 3.0进行蛋白质结构预测和寡聚化模拟;构建了CRISPR抗性CLCC1突变体进行功能拯救实验。
研究人员通过全基因组CRISPR筛选发现,在HSV-1感染的HeLa细胞中,CLCC1缺失导致核逃逸效率降低约80%。流式细胞术分析和共聚焦显微镜观察均证实,CLCC1-KO细胞中细胞质衣壳信号显著减少,表明核逃逸过程在融合阶段受阻。
除HSV-1外,CLCC1缺失同样显著抑制HSV-2和PRV的复制,病毒滴度下降约1000倍。系统进化分析显示,CLCC1同源基因在感染软体动物和鱼类的疱疹病毒基因组中存在,这些vCLCC1通过三次独立的水平基因转移事件从宿主基因组获得,表明CLCC1功能在疱疹病毒目中的普遍重要性。
在未感染细胞中,CLCC1缺失引起核膜出泡(NBs)现象,核膜上出现直径80-260纳米的球形囊泡。这些NBs沿核膜呈"串珠状"分布,部分通过颈部与INM相连,形态类似于Torsin ATP酶缺失表型,表明CLCC1在核孔复合体插入或核输出中发挥作用。
虽然CLCC1此前与ER应激相关,但研究发现HSV-1感染期间,ER应激标志物BiP水平较低,且ER应激诱导剂二硫苏糖醇(DTT)不影响核逃逸效率,表明CLCC1在核逃逸中的作用独立于ER应激通路。
AlphaFold结构预测显示,CLCC1核心区域(残基161-360)在病毒和细胞同源蛋白中高度保守。关键残基突变实验表明,TM2结构域(S224A)和拳状结构域(C254A、C254A/C279A、D277R)的突变破坏CLCC1功能,而通道活性相关突变影响较小,提示CLCC1可能通过直接参与膜融合而非离子通道功能发挥作用。
本研究首次揭示了CLCC1在疱疹病毒核逃逸中的关键作用,建立了病毒复制与细胞核膜融合机制之间的直接联系。研究发现不仅阐明了疱疹病毒利用宿主因子完成核膜融合的分子机制,还揭示了CLCC1在细胞核孔复合体形成等基本生物学过程中的功能。
从进化角度看,疱疹病毒多次独立获得CLCC1同源基因的现象强烈表明,这一宿主因子对病毒复制的普遍重要性。病毒可能通过水平基因转移"窃取"宿主CLCC1基因,以确保在感染过程中更精确地调控核膜融合过程。
从机制层面,研究提出了CLCC1可能通过其拳状结构域与靶膜互作,通过寡聚化诱导膜弯曲,最终促进膜融合的工作模型。CLCC1在ER膜上的定位使其能够直接参与INM和ONM的融合过程,这一机制可能普遍适用于多种核膜相关过程。
该研究的发现为开发广谱抗疱疹病毒策略提供了新靶点。鉴于CLCC1在多种疱疹病毒核逃逸中的保守作用,针对CLCC1的抑制剂可能对包括HSV-1、HSV-2、PRV等在内的多种疱疹病毒具有抑制作用。同时,对CLCC1功能的深入理解也有助于阐明核孔复合体生物发生、核膜出芽等基本细胞过程的分子机制。
然而,研究也存在一定局限性,如CLCC1是否直接具有融合活性仍需实验验证,以及核膜出泡现象是否完全由核孔复合体形成缺陷引起尚需进一步研究。未来工作将聚焦于解析CLCC1的精确工作机制及其在病毒与宿主互作中的全面功能。
本研究由美国塔夫茨大学医学院Ekaterina E. Heldwein团队主导,发表于《Nature Communications》期刊,为理解疱疹病毒核逃逸机制和核膜融合过程提供了重要突破,为开发新型抗病毒策略奠定了理论基础。
 生物通微信公众号
生物通微信公众号
 生物通新浪微博
生物通新浪微博
今日动态 |
人才市场 |
新技术专栏 |
中国科学人 |
云展台 |
BioHot |
云讲堂直播 |
会展中心 |
特价专栏 |
技术快讯 |
免费试用
版权所有 生物通
Copyright© eBiotrade.com, All Rights Reserved
联系信箱:
粤ICP备09063491号